

Batch Manufacturing Record (BMR)Glossary
Batch Manufacturing Record Guide
This topic is part of the SG Systems Global regulatory, MES and process manufacturing glossary.
Updated January 2026 • Batch Documentation, Master Manufacturing Record (MMR), Master Batch Record (MBR), Electronic Batch Record (eBR), Weighing & Dispensing Control, MES / WMS / ERP Integration • Pharma, Biologics, API, Food, Nutrition, Cosmetics, Chemicals
A Batch Manufacturing Record (BMR) is the definitive, batch-specific record of how a product was actually made – not how it was supposed to be made. It starts from the instructions in the Master Manufacturing Record (MMR) or Master Batch Record (MBR), then documents the real-world details: which lots were used, which equipment ran, who did what, when, at what settings, with what results, and what went wrong (or right) along the way. Regulators, customers and internal QA teams all use BMRs to answer one core question: “Can you prove this batch was made in a controlled, compliant way – and would you stand behind it in court?”
“If your batch manufacturing records are weak, every batch release is an act of faith – and every investigation becomes guesswork.”
TL;DR: A Batch Manufacturing Record (BMR) is the batch-level execution record derived from a master recipe (MMR / MBR). It captures materials, equipment, line settings, step-by-step operations, in-process controls, QC tests, deviations, yields, packaging and approvals for a single batch. In a modern plant, BMRs are implemented as electronic batch records (eBR) orchestrated by MES, integrated with weighing & dispensing, WMS and ERP. Good BMRs are structured, link-rich, and ALCOA+ compliant; they drive release, recalls, investigations, continuous improvement and digital transformation. Bad BMRs are loose paper piles, spreadsheet islands and tribal knowledge – and they silently erode your ability to defend your product.
1) What is a Batch Manufacturing Record (BMR)?
A Batch Manufacturing Record is the written or electronic record that documents the full manufacturing history of a specific batch or lot – from material staging and weigh-and-dispense through processing, filling, packaging, and release. It is sometimes called a Batch Production Record (BPR) in some sectors, but the structure and intent are similar:
- MMR/MBR = “recipe and rules” – defines what should happen in each step.
- BMR = “batch story” – records what did happen for this specific batch.
In regulated industries, BMRs are mandated by authorities:
- FDA drug and biologics cGMPs (21 CFR 211) – require batch production and control records.
- Dietary supplements (21 CFR 111) and food (21 CFR 117) – require production records that demonstrate control over critical parameters.
- EU GMPs and PIC/S PE 009 – require comprehensive batch documentation and Quality Assurance review.
- GFSI schemes (e.g., BRCGS, SQF, FSSC) – expect structured batch records as part of QMS and traceability.
Even where strict GMPs do not apply (e.g., many chemical or industrial process environments), BMR-style documentation is increasingly demanded by customers and certification schemes as a pillar of robust manufacturing governance.
2) BMR vs. MMR / MBR / MPR / BPR – How They All Fit Together
The acronym forest gets dense quickly, so let’s anchor the relationships clearly:
- MMR (Master Manufacturing Record) – authorised master instructions for manufacturing a given product.
- MBR (Master Batch Record) – often synonymous with MMR in pharma/biologics; may include more process detail for batch-based systems.
- MPR (Master Packaging Record) – master instructions for packaging bulk material into final presentation (tablets into blisters, bulk cream into tubes, etc.).
- BPR (Batch Packaging Record) – batch-level packaging execution record, analogous to BMR but focused on packaging operations and labelling control.
- BMR (Batch Manufacturing Record) – batch-level manufacturing execution record for this production order or lot.
- eBR (Electronic Batch Record) – digital implementation of BMR/BPR inside an MES or ERP environment.
A typical data flow:
- Product & process development define the MMR/MBR and MPR; QA approves them under change control.
- ERP generates a production order; MES instantiates a batch-level BMR from the master.
- Operators execute the batch under MES guidance; BMR/eBR captures actions, data and deviations.
- Packaging executes under MPR/BPR; final disposition combines BMR+BPR into a full “batch history.”
Getting this relationship tight – especially between MMR/MBR and BMR/eBR – is critical. Many inspection findings and customer complaints trace back to mismatches between “what the master says” and “what the batch record shows actually happened.”
3) Regulatory & Scheme Expectations for BMRs
Different regulators use different language, but the core BMR expectations are consistent: completeness, traceability and reviewability.
- 21 CFR 211 (pharma) – requires production and control records for each batch, including component lots, processing instructions, in-process control results, yields, lab tests, investigation outcomes and signatures.
- 21 CFR 111 (supplements) – demands batch production records that document each significant manufacturing step and link to the master, including component control and deviations.
- 21 CFR 117 (food) – requires records showing implementation of preventive controls, often naturally structured into BMR-like documents in process-focused operations.
- EU GMP / PIC/S – emphasise batch records as the main evidence for QP release, including cross-links to equipment logs, cleaning, calibration and environment.
- GFSI schemes – BRCGS, SQF, FSSC, etc. – require documented process control, traceability, mock recalls and CAPA, all strongly dependent on good BMR data.
The message is simple: if you can’t show it in the batch record, regulators and customers will assume it didn’t happen – or at least that you can’t prove it did.
4) Essential Content of a BMR – A Practical Checklist
While every industry has nuances, a robust BMR template usually covers:
- Header & identification
- Product name, strength/grade, internal code.
- Batch/lot number, order number, campaign link.
- Reference MMR/MBR number and version.
- Target batch size and units.
- Material usage (weighing & dispensing)
- Material names and codes; supplier; CoA references.
- Lot numbers, expiry/retest dates, quantities (target vs actual) and tolerances.
- Sign-offs for weighed/dispensed materials and second-person verification where required.
- Equipment & line assignment
- Equipment IDs (tanks, mixers, lines, PLCs), cleanliness status, calibration status.
- Line clearance records (line clearance), pre-use checks.
- Step-by-step manufacturing instructions & data
- Sequence of operations with targets for time, temperature, speed, pH, pressure, etc.
- Fields for actual values, start/finish times, operator IDs, and comments.
- Embedded in-process control (IPC) checks.
- Quality control & testing
- In-process and finished-product tests, methods, specs and results.
- Pass/fail status for each parameter; linkage to LIMS sample IDs.
- Yield and reconciliation
- Theoretical vs actual yields; scrap; rework; mass-balance closing.
- Byproduct/co-product identification, amounts and disposition.
- Deviations / nonconformances
- Final disposition & approvals
- Batch status (Released / Rejected / Quarantined / Reworked); QA/QP signatures and dates.
That sounds like a lot – because it is. That’s why BMRs are a natural focal point for digitalisation and MES-driven execution: you want all that richness, but you want it captured automatically and accurately, not via midnight handwriting marathons.
5) BMR as a Central Node in Your QMS
BMRs sit at the intersection of:
- Manufacturing – what actually happened on the line.
- Quality Control – what was measured and whether it met the spec.
- Quality Assurance – whether the batch, and the system, behaved as intended.
- Regulatory Compliance – ability to demonstrate adherence to GMP, FSMA, or GFSI expectations.
- Business Performance – yields, scrap, cycle times, OEE, complaint rates.
When BMRs are treated only as a regulatory overhead, organisations leave a lot of value on the table. When they’re treated as the core data layer of the QMS, they fuel smarter decision-making, faster root cause analysis, leaner audits and clearer risk management. This is especially true once they’re electronic and integrated with process historians, SPC and OEE analytics.
6) How BMRs Connect to MES, ERP and WMS
In a fully integrated environment, the BMR is not a static document but a live manifestation of data flows:
- ERP sends the production order with required product, quantity, due date and BOM; this spawns a BMR instance in MES.
- WMS manages goods receipt (GS1-128 intake capture), lot transfers and batch-to-bin traceability; picking and staging are recorded via scanning and feed into the BMR material section.
- MES orchestrates execution – step-by-step instructions, data capture from equipment, EWIs, in-process checks, electronic signatures – populating the BMR/eBR in real time.
- LIMS manages QC testing and sends results back into the BMR; if integrated, CoAs and release decisions are generated automatically.
- ERP/WMS gets final yield and disposition data; finished-goods lots, shipments and invoices are all traceable back to the BMR.
Well-designed BMR/eBR implementations hide most of this complexity from the operator. They see a guided workflow; the system takes care of wiring data to the right objects in the background, ensuring that the BMR is complete and accurate without requiring them to think like a data architect.
7) Handling Scaling, UoM, Loss and Co-Products in BMRs
For process manufacturers, scaling and yield management are central pain points that BMRs can either clarify or obscure. Good practice:
- Embed scaling logic via dynamic recipe scaling so that the BMR shows scaled targets automatically when planners adjust batch size.
- Manage unit-of-measure conversions centrally and apply them consistently in BMRs, ERP and WMS to avoid conflicting quantities.
- Distinguish expected losses (filter solids, line hold-up) from unexpected scrap and track both in BMR yield sections.
- Capture co-products and byproducts explicitly, tying them into traceability so they can be followed downstream through packaging and customer shipments.
When this is done properly, a BMR can fuel accurate mass-balance, cost-to-serve and sustainability metrics – making it easier to justify process improvements or new equipment based on real, quantified loss patterns rather than intuition alone.
8) A Visual Example – eBMR with Recipe, BOM, Allergens & Warnings
Sometimes a picture is worth a thousand bullet points. The example below shows a sample eBMR screen from an SG Systems-style implementation, combining recipe, BOM, allergen flags and warnings in a single operator-facing view:
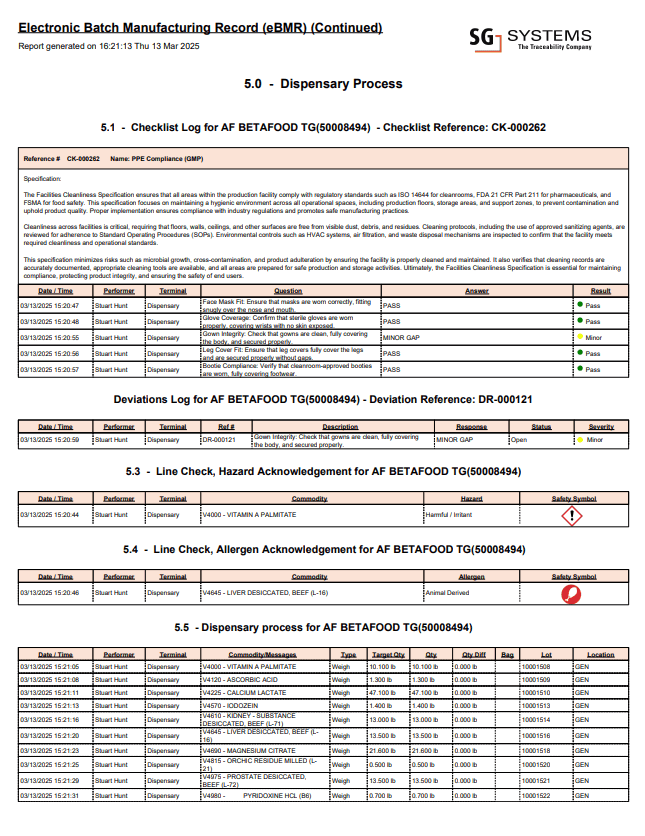
This kind of design makes the BMR not just a record but a real-time guide: operators see exactly what material to add, where it is staged, which allergens and hazards are involved, what PPE is required, and what limits must be met – with everything captured back into the eBR automatically. For auditors, having the UI and the underlying record aligned reduces arguments about “what the SOP says” vs “what the screen tells people to do.”
9) Paper BMR vs eBR – Data Integrity and ALCOA+
Traditional paper BMRs suffer from predictable problems: missing data, illegible handwriting, back-dating, lost attachments and limited searchability. In contrast, a well-implemented eBR supports ALCOA+ by design:
- Attributable – each entry tied to a unique user ID with exact timestamps.
- Legible – consistent formatting, no penmanship issues.
- Contemporaneous – tasks and data points created in real time, not end-of-shift.
- Original – data originates in the system, often directly from equipment.
- Accurate – automatic checks, equipment interfaces and hard-gating reduce corrections.
- Complete, Consistent, Enduring, Available – extra ALCOA+ principles that eBRs support through structured storage and robust access controls.
Regulators and customers are increasingly impatient with batch records that look like archaeological artefacts. Moving BMRs into a validated eBR platform is one of the single most impactful steps a plant can take to reduce compliance risk and investigation complexity – especially when tied into OEE, historians and other digital initiatives.
10) BMR Workflows – Routing, Review and Approval
Whether on paper or in eBR, BMRs must follow a disciplined workflow:
- Creation – from an approved MMR/MBR, automatically pulled into the batch instance.
- Execution – operators complete steps, capture data and sign off; MES enforces sequence and necessary checks.
- Supervisor review – line or area managers review for completeness, obvious errors and unresolved exceptions.
- QA/QP review – quality reviews all critical data, deviations and investigations; then approves or rejects the batch.
- Archival – BMR is archived per retention rules and remains accessible for audits, investigations and PQR.
In a mature environment, this workflow is clearly documented in SOPs and embedded into the eBR/MES system so users are guided and blocked at the right times. Integration with quality agreements and MOC ensures that changes to the master are properly propagated into new BMR templates and that historical BMRs remain traceable to the masters under which they were produced.
11) What Good BMR Software Should Do – Beyond Form Filling
“BMR software” that simply mimics forms misses most of the opportunity. In process manufacturing, look for solutions that offer:
- Contextual guidance – step-by-step, operator-level guidance with visual cues, hazard warnings, allergen flags and PPE requirements.
- Deep integration – with scales, PLCs, load cells, LIMS, WMS and ERP.
- Industry-specific features – formulation/recipe support, solids/assay corrections, potency adjustments, multiple phases, heating/cooling profiles.
- Compliance automation – built-in support for data integrity, 21 CFR 11, EU Annex 11, and GFSI-aligned recordkeeping.
- Analytics readiness – structured data models that make it easy to query BMR data across batches for SPC, yield and CAPA analysis.
The distinguishing mark of good BMR/eBR software is that it reduces batch-cycle friction while improving data richness, instead of simply moving paperwork from clipboards into tablets without changing workflow logic.
12) Common BMR Pitfalls (and How to Avoid Them)
In audits and remediation projects, the same BMR pain points appear repeatedly:
- Incomplete or inconsistent records – missing fields, sections or attachments; inconsistent use of templates across batches.
- Backdating and reconstructed entries – undermining credibility and violating ALCOA principles.
- Unclear link to master – BMRs that do not clearly reference the correct MMR/MBR version or reflect its instructions.
- Fragmented data – process data, QC data and deviations living in separate systems with weak cross-references.
- Complexity overload – trying to capture everything in the BMR, making it unusable for operators and reviewers.
Avoidance strategies: standardise templates and governance, embed BMR logic in MES rather than on paper, use internal audits and mock inspections, and treat BMR design as part of your broader QMS and digital roadmap, not as a standalone documentation “project.”
13) BMRs, Investigations and CAPA
When something goes wrong, the BMR is where you start:
- Complaints and adverse events – mapping suspect lots back to BMRs and then to MMR/MBR, materials, equipment and process history.
- OOS/OOT results – using BMR data to identify patterns: parameter drifts, atypical conditions, operator changes.
- EM and hygiene issues – cross-referencing cleaning records, line clearance, and batch sequences in BMRs.
- Regulatory or customer inquiries – providing documented evidence for each batch under question.
CAPA effectiveness depends heavily on BMR quality. If BMRs are incomplete or inconsistent, your root cause analysis is based on guesswork. If they are structured and integrated, you can rapidly identify which factors changed when, and whether CAPA measures actually produce sustained improvements in subsequent BMRs – closing the loop between documentation and real-world control.
14) Designing a BMR Template – Practical Tips
A well-designed Batch Manufacturing Record template makes it easier for operators to execute consistently, for reviewers to understand what happened, and for auditors to trace decisions. Practical considerations include:
- Base the layout on the actual process flow – sections should follow the real manufacturing sequence (staging → weighing → processing → packaging), not departmental silos.
- Use consistent terminology and IDs – step names, equipment codes and test names should match those used in the MMR / MBR, SOPs, HMI screens and training materials.
- Prioritise critical data – highlight fields that are essential for batch release (materials, critical parameters, IPCs, QC results, deviations) and avoid cluttering the template with low-value information.
- Minimise free-text fields – where possible, use structured fields, dropdowns and coded entries instead of long comment boxes to improve data quality and enable analysis.
- Embed references – include MMR/MBR numbers, SOP IDs, test method references and links to relevant specifications so that reviewers and auditors can quickly find supporting documents.
- Design for review and investigation – group information so that a reviewer can easily move from header information to materials, steps, tests, yields, deviations and final disposition without paging back and forth.
- Plan for scalability – use a template structure that can support additional products, lines and regulatory expectations over time without needing to be redesigned from scratch.
For electronic batch records, these principles apply both to the underlying data model and to the user interface presented to operators. Clear, logical templates reduce errors, speed up batch review and make it easier to demonstrate control to regulators, customers and internal stakeholders.
15) FAQ
Q1. What is the difference between an MMR/MBR and a BMR?
The MMR / MBR is the master “recipe and rules” for making a product. It defines materials, equipment, steps, process parameters, controls and limits. The BMR is the batch-specific record showing how those instructions were actually applied for a given batch, including actual values, deviations, tests and approvals. You need both: a sound master and a complete, accurate batch history.
Q2. Do Batch Manufacturing Records have to be electronic (eBR)?
No. Paper BMRs can be compliant if they meet regulatory expectations for completeness, legibility, contemporaneity, control and retention. However, electronic batch records (eBR) implemented in an MES or ERP-integrated environment make it much easier to achieve data integrity (ALCOA+), integrate with equipment and other systems, reduce manual errors and respond quickly to audits and investigations.
Q3. How detailed does a BMR need to be?
It must be detailed enough for a competent reviewer or inspector to reconstruct exactly how the batch was manufactured, verify that all critical parameters and tests met specifications, and understand the impact of any deviations. That means including material lots, equipment IDs, real-time process values, in-process and final test results, yields, deviations and final disposition – not just a batch number, a date and “OK” tick boxes.
Q4. How are BMRs used in investigations and CAPA?
BMRs are the primary evidence source when investigating complaints, OOS/OOT results, deviations, environmental or hygiene issues. Investigators examine BMRs to see whether the batch followed the approved MMR/MBR, where it deviated, whether similar deviations occurred in other batches, and whether previous CAPAs were effective. High-quality, consistent BMR data significantly shorten investigations and strengthen the credibility of CAPA conclusions.
Q5. Where should we start if our BMRs are mostly manual, inconsistent and hard to review?
Start by defining a standard BMR structure that aligns with your MMR/MBR, SOPs and regulatory/scheme expectations. Pilot a cleaned-up template on one high-risk or high-volume product family, focusing on clarity, completeness and alignment with real workflows. Then plan a phased move towards eBR within an MES or ERP-integrated solution, prioritising lines with the highest audit pressure. Use internal audits and mock regulator/customer reviews to refine the design before scaling – and treat BMR improvement as part of your broader QMS and digital-transformation roadmap, not as a one-off documentation clean-up.
Related Reading
• Master & Batch Records: Master Manufacturing Record (MMR) | Master Batch Record (MBR) | Electronic Batch Record (eBR)
• Execution & Systems: Weighing & Dispensing Component Control | Weigh-and-Dispense Automation | MES | WMS | ERP
• Quality, Risk & Compliance: QMS | Certificate of Analysis (CoA) | Deviation / NC | CAPA | Risk Management (QRM) | Data Integrity
• Traceability & Analytics: End-to-End Lot Genealogy | Mass Balance | Yield Variance | Product Quality Review (PQR)
OUR SOLUTIONS
Three Systems. One Seamless Experience.
Explore how V5 MES, QMS, and WMS work together to digitize production, automate compliance, and track inventory — all without the paperwork.

Manufacturing Execution (MES)
Control every batch, every step.
Direct every batch, blend, and product with live workflows, spec enforcement, deviation tracking, and batch review—no clipboards needed.
- Faster batch cycles
- Error-proof production
- Full electronic traceability

Quality Management (QMS)
Enforce quality, not paperwork.
Capture every SOP, check, and audit with real-time compliance, deviation control, CAPA workflows, and digital signatures—no binders needed.
- 100% paperless compliance
- Instant deviation alerts
- Audit-ready, always

Warehouse Management (WMS)
Inventory you can trust.
Track every bag, batch, and pallet with live inventory, allergen segregation, expiry control, and automated labeling.
- Full lot and expiry traceability
- FEFO/FIFO enforced
- Real-time stock accuracy
You're in great company



















How can we help you today?
We’re ready when you are.
Choose your path below — whether you're looking for a free trial, a live demo, or a customized setup, our team will guide you through every step.
Let’s get started — fill out the quick form below.